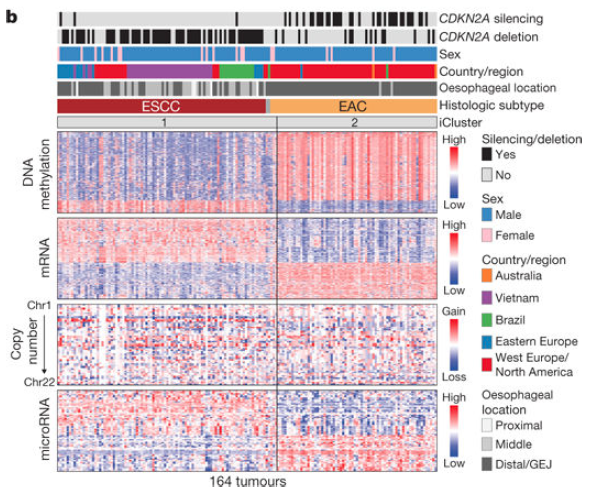
高级部分
参数选择: 上面的 iClusterPlus设置的K和lambda其实需要很多次调整。要用到 tune.iClusterPlus对K值进行多次遍历, 之后从中选择最优K值和lamda值。
如下分析推荐用Linux服务器进行, 否则速度会比较慢(Windows用户的cpus只能为1),具体时间视数据量大小而定
如下面的代码就跑了差不多10个小时, 平均每个循环2小时, Rdata已经保存。
set.seed(123) # 随机数种子保证每次结果一致性
before_run <- Sys.Date()
for(k in 1:5){
file_name <- paste("cv.fit.k",k,".Rdata",sep="")
if ( ! file.exists(file_name)){
cv.fit <- tune.iClusterPlus(cpus=15,dt1=gbm.mut2,dt2=gbm.cn,dt3=gbm.exp,
type=c("binomial","gaussian","gaussian"),
K=k,n.lambda=185,
scale.lambda=c(1,1,1),maxiter=20)
save(cv.fit, file=file_name) #保存到Rdata文件里,便于中断后继续
}
}
after_run <- Sys.Date()
paste0("Running time is ", after_run-before_run)
这里的"K1=5"是主要的遍历参数, 而lambda根据使用数据集数目选择, 参考下表. 当然实在不会选就用 lambda=NULL用默认值。
x1 <- c(1,2,3,4)
x2 <- c("任意选择",
"8, 13, 21, 34, 55, 89, 144, 233, 377, 610",
"35, 101, 135, 185, 266, 418, 579, 828, 1010",
"307, 526, 701, 1019, 2129, 3001, 4001, 5003, 6007")
df <- data.frame(x1 = x1, x2=x2)
colnames(df) <- c("数据类型种类","lambda选择值")
knitr::kable(df, format = "html")
遍历完成之后, 就可以从结果中选择合适的参数。
#读取保存的Rdata
output <- alist() # alist里的参数惰性求值
files <- grep("cv.fit",dir())
for(i in 1:length(files)){
load(dir()[files[i]])
output[[i]] <- cv.fit
}
# 提取之前设置的K和Lambda
nLambda <- nrow(output[[1]]$lambda)
nK <- length(output)
# 获取每个拟合结果中的BIC值
BIC <- getBIC(output)
# 获取每个拟合结果中的
devR <- getDevR(output)
# 寻找每个K中最小BIC对应lambda ID
minBICid <- apply(BIC,2,which.min)
# 计算最小BIC的
devRatMinBIC <- rep(NA,nK)
for(i in 1:nK){
devRatMinBIC[i] <- devR[minBICid[i],i]
}
plot(1:(nK+1),c(0,devRatMinBIC),
type="b",
xlab="Number of clusters (K+1)",
ylab="%Explained Variation")
我们主要根据偏差率(deviance ratio)选择最优K, 计算方式不是重点, 重点是作图后下图里第一个到顶对应的K. (上图K+1=3,所以K=2)。
根据上图结果,从多次遍历中提取最优的聚类和拟合
clusters <- getClusters(output)
rownames(clusters) <- rownames(gbm.exp)
colnames(clusters) <- paste("K=",2:(length(output)+1),sep="")
head(clusters)
#保存clusters到文件
#write.table(clusters, file="output_clusterMembership.txt",sep='\t',quote=F)
output_clusterMembership.txt文件作为第一个输出文件,选一列分类,作为画热图时的sample分组信息。
k <- 2
best.cluster <- clusters[,k]
best.fit <- output[[k]]$fit[[which.min(BIC[,k])]]
特征选择: 对上面找到的3个聚类(K=2),可以根据lassso系数估计选择最高的几个特征
features <- alist()
features[[1]] <- colnames(gbm.mut2)
features[[2]] <- colnames(gbm.cn)
features[[3]] <- colnames(gbm.exp)
sigfeatures=alist()
for(i in 1:3){
rowsum <- apply(abs(best.fit$beta[[i]]),1, sum)
upper <- quantile(rowsum,prob=0.75)
sigfeatures[[i]] <- (features[[i]])[which(rowsum>upper)]
}
names(sigfeatures) <- c("mutation","copy number","expression")
#输出每个数据集的前几个特征
head(sigfeatures[[1]])
head(sigfeatures[[2]])
head(sigfeatures[[3]])
如果你发现某一个数据集没有显著的特征,那你就需要调整“高级分析”第一段 tune.iClusterPlus函数中的 scale.lambda, 比如说设置成 c(0.05,1,1)然后重新跑。
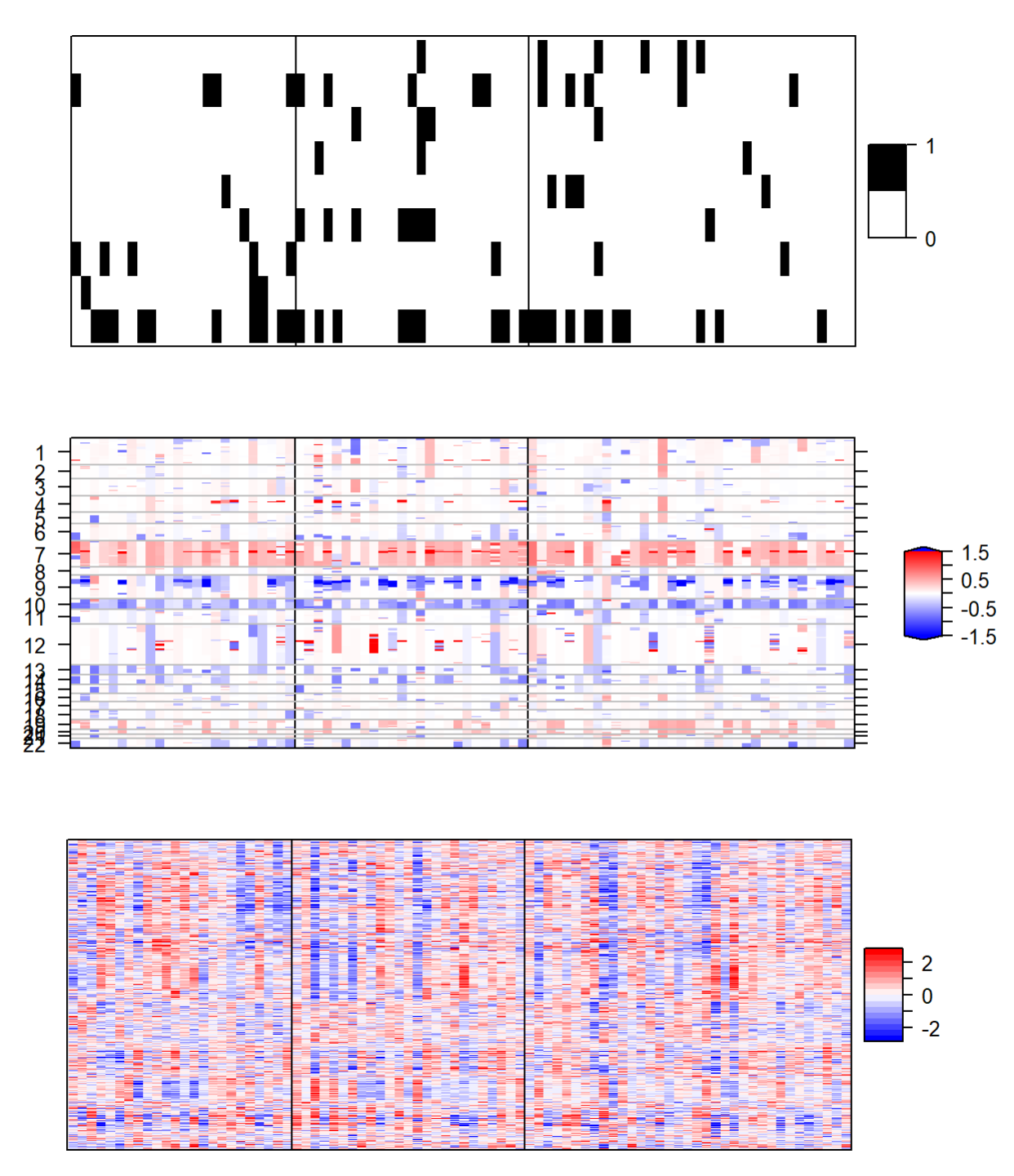
热图展示: 基于最优拟合画热图
library(magrittr)
# 提取gbm.cn的染色体位置信息
chr <- unlist(strsplit(colnames(gbm.cn), '\\.')) %>%
extract(seq(1,length(.),by=2)) %>%
gsub("chr","",.) %>%
as.numeric()
# 为了更好的展示对过高的值调整
cn.image <- gbm.cn
cn.image[cn.image > 1.5] <- 1.5
cn.image[cn.image < -1.5] <- -1.5
exp.image <- gbm.exp
exp.image[exp.image > 2.5] <- 2.5
exp.image[exp.image < -2.5] <- -2.5
# 选择配色
bw.col <- colorpanel(2,low="white",high="black")
col.scheme = alist()
col.scheme[[1]] <- bw.col
col.scheme[[2]] <- bluered(256)
col.scheme[[3]] <- bluered(256)
# 作图
plotHeatmap(fit=best.fit,datasets=list(gbm.mut2,cn.image,exp.image),
type=c("binomial","gaussian","gaussian"),
col.scheme = col.scheme,
row.order=c(F,F,T),chr=chr,plot.chr=c(F,T,F),
sparse=c(T,F,T),cap=c(F,T,F))
输出文件
把clusters、特征基因和值保存到文件,就可以用自己的heatmap代码画热图了。
write.table(clusters, file="output_clusterMembership.txt",sep='\t',quote=F)
write.csv(t(gbm.mut2),"output_mut2.csv",quote = F)
write.csv(t(cn.image),"output_cn.csv",quote = F)
write.csv(t(exp.image),"output_exp.csv",quote = F)
 发表于 2025-6-2 09:51:54
发表于 2025-6-2 09:51:54
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )