背景
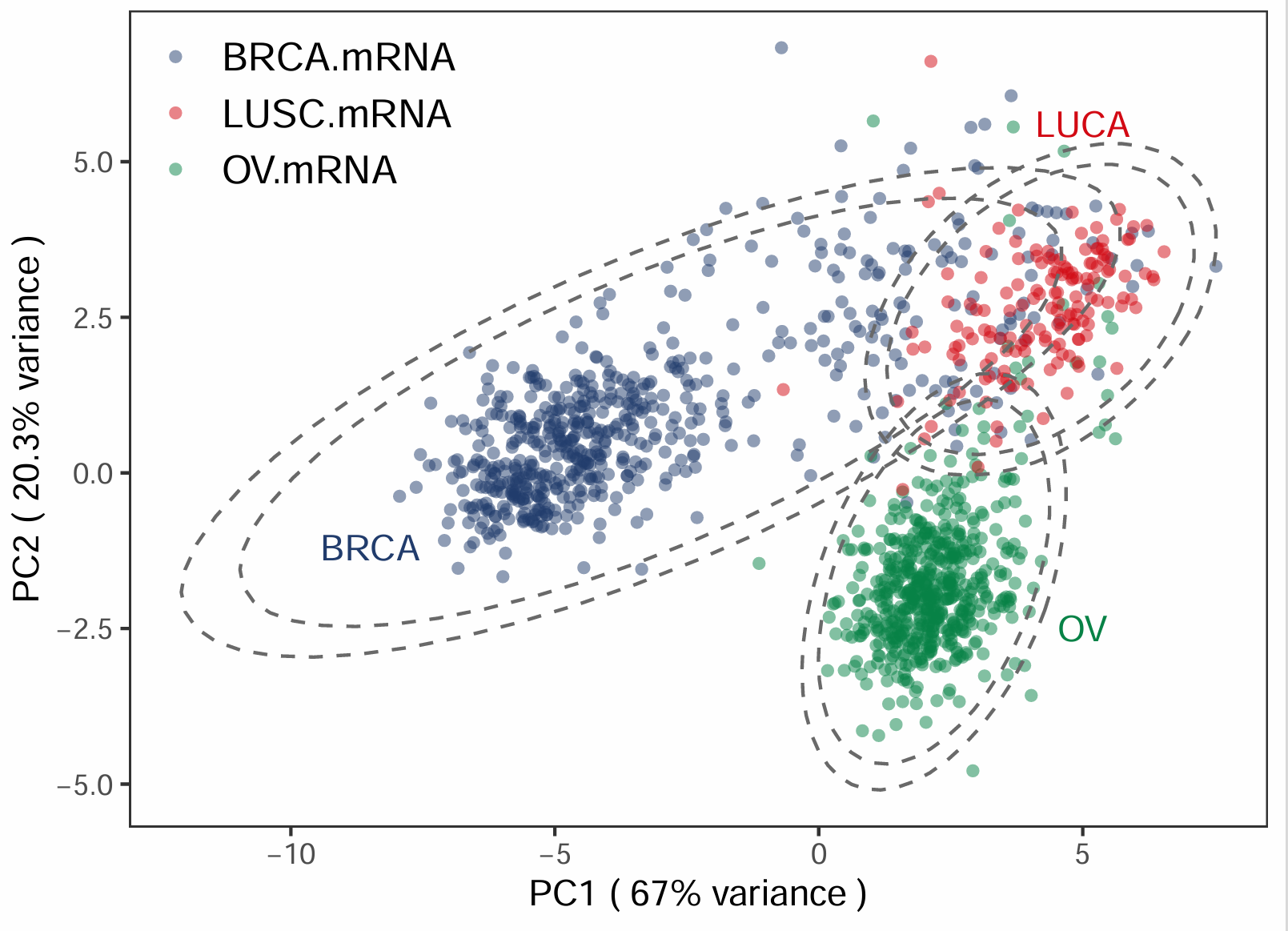
用TCGA数据画主成分分析(pricinple components analysis,PCA)的图。
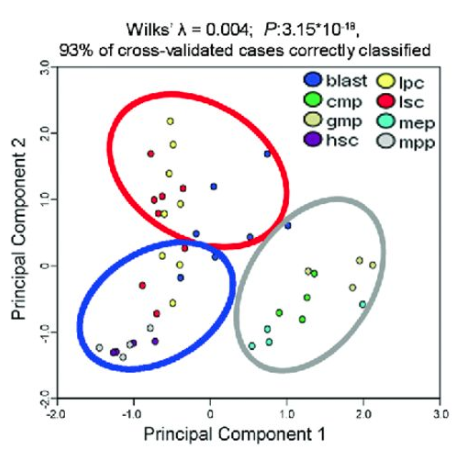
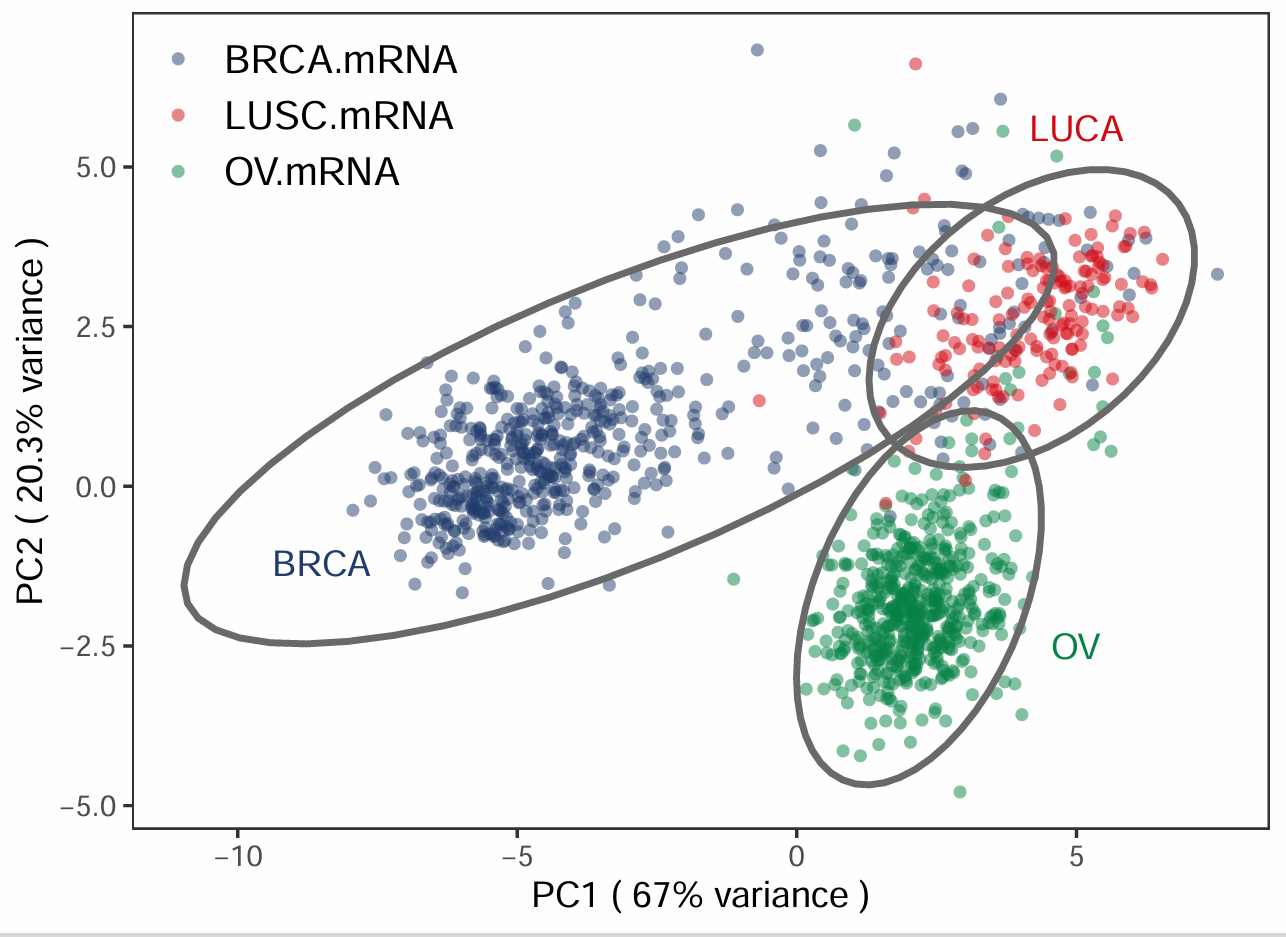
出自文章PMID: 28411251
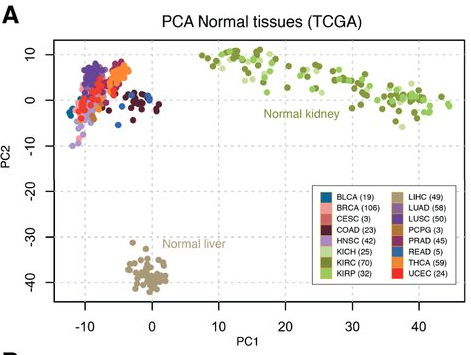
出自文章PMID: 27663598
应用场景
通常我们拿到的数据集,例如转录组的表达量矩阵都有成千上万个基因,这类高维度数据很难直接进行可视化展示。
为了能够在二维平面上展示,就需要对其进行降维,而PCA分析是目前最常用的降维工具。
然后你就能在图上一眼看出哪些样品之间更像,哪些样品属于/不属于同一组。不仅适用于测序数据。
场景一:多个分组,每组三次生物学重复。做PCA,看到某个样品远离了组内其他样品,它有什么问题?
场景二:多个分组,分三批送去测序。做PCA,看不出分组的规律,却看到同一批次的样品间距离更近,批次效应?
输入数据
两个输入文件:
- 表达量矩阵:easy_input_expr.csv,第一列是样品名,后续几列是各个基因的表达量,或者其他属性
- 样品分组:easy_input_meta.csv,第一列是样品名,第二列是分组
此处输入数据为3种癌症BRCA、OV、LUSC里,5个基因"GATA3", "PTEN", "XBP1","ESR1", "MUC1"的表达量。
数据来源:用R包 RTCGA.mRNA提取TCGA数据。
expr_df <- read.csv(file='easy_input_expr.csv',row.names = 1,
header = TRUE, sep=",", stringsAsFactors = FALSE)
meta_df <- read.csv(file='easy_input_meta.csv', row.names = 1,
header = TRUE, sep=",",stringsAsFactors = FALSE)
#查看前3个基因在前4个sample中的表达矩阵
expr_df[1:3,1:4]
#查看样本信息前3行
head(meta_df, n=3)
做PCA
#用`prcomp`进行PCA分析
pca.results <- prcomp(expr_df, center = TRUE, scale. = FALSE)
#定义足够多的颜色,用于展示分组
mycol <- c("#223D6C","#D20A13","#088247","#FFD121","#11AA4D","#58CDD9","#7A142C","#5D90BA","#431A3D","#91612D","#6E568C","#E0367A","#D8D155","#64495D","#7CC767")
开始画图
这里提供两种方法:
- 方法一:自动画图。用现成的R包全自动画图,必要时只需调整参数。
- 方法二:提取PCA分析结果,手动画图。
方法一:自动画图
用到做PCA的R包 ggord,优点是能用背景色展示置信区间,缺点是没有画圈功能。
Y叔对ggord进行了加强,放在R包 yyplot里,yyplot包里提供的 geom_ord_ellipse能画出多个圆圈,用来展示不同的置信区间,并且可以反复叠加,想画几个圈就画几个圈。
经典版
#install.packages("devtools")
#library(devtools)
#devtools::install_github("GuangchuangYu/yyplot")
#devtools::install_github('fawda123/ggord')
library(ggplot2)
library(plyr)
library(ggord)
#library(yyplot)
#有可能你的网络在安装yyplot时遇到困难,我把geom_ord_ellipse函数单独下载,通过本地进行加载。
#调用yyplot包里的geom_ord_ellipse函数
source('./geom_ord_ellipse.R') #该文件位于当前文件夹
p <- ggord(pca.results, grp_in = meta_df$group, repel=TRUE,
ellipse = FALSE, #不显示置信区间背景色
size = 2, #样本的点大小
alpha=0.5, #设置点为半透明,出现叠加的效果
#如果用自定义的颜色,就运行下面这行
cols = mycol[1:length(unique(meta_df$group))],
arrow = NULL,txt = NULL) + #不画箭头和箭头上的文字
theme(panel.grid =element_blank())
p +
geom_ord_ellipse(
ellipse_pro = 0.95,
size = 1.5,
lty = 1
)
#用ggord画基本PCA图
#保存到pdf文件
ggsave("PCA_classic.pdf", width = 6, height = 6)
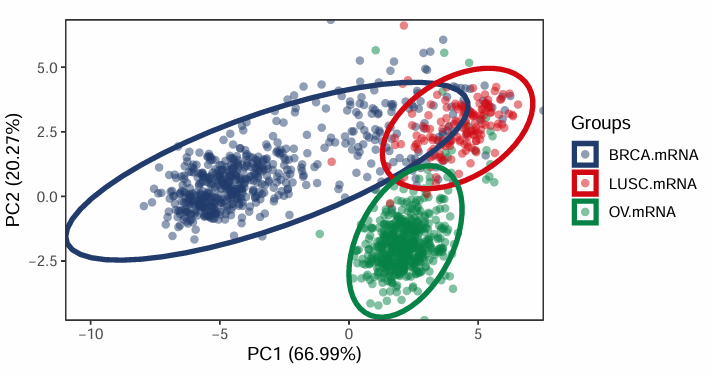
箭头版
#用ggord画基本PCA图和置信区间背景色
ggord(pca.results, grp_in = meta_df$group, repel=TRUE,
alpha = 0.6,#点和置信区间背景设为半透明,以凸显箭头和文字
#或者单独修改置信区间背景的透明度
#alpha_el = 0.3,
ellipse_pro = 0.95,#置信区间
size = 2,
#如果想用默认的颜色,就在下面这行前面加个#
#cols = mycol[1:length(unique(meta_df$group))],
arrow=0.2, #箭头的头的大小
vec_ext = 5,#箭头尾巴长短
veccol="brown",#箭头颜色
txt=3) + #箭头指向的基因名的字体大小
theme(panel.grid =element_blank()) +
#用yyplot继续添加虚线的置信区间
geom_ord_ellipse(ellipse_pro = .95, #先画个.95的圆圈
color='darkgrey', #圈圈的颜色
size=0.5,
lty=2 ) + #画成虚线,可以用1-6的数字设置为其他线型
geom_ord_ellipse(ellipse_pro = .98, #再画个.98的圆圈
#color='grey', #把这行注释掉,就是跟点一样的颜色
size=0.5, lty=2 )
#保存到pdf文件
ggsave("PCA_arrow.pdf", width = 6, height = 6)
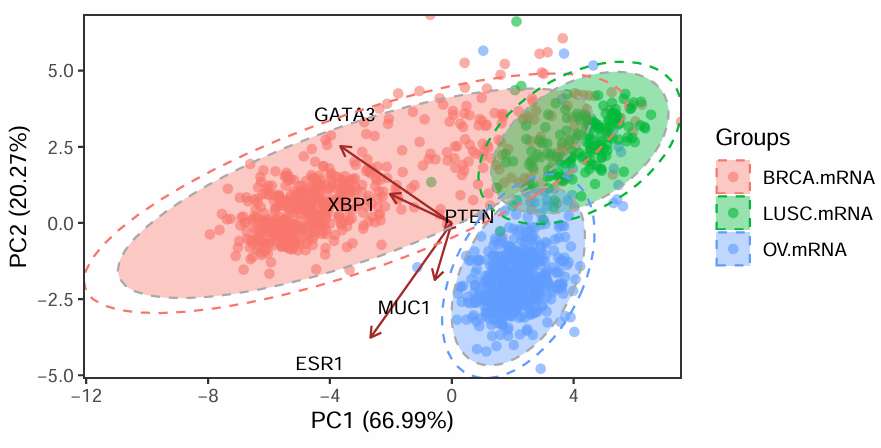
方法二:提取PCA分析结果,手动画图
从PCA结果到画图所需的输入数据整理
用 prcomp进行PCA分析后,获取降维后每个样本对应的主成分值和每个主成分的解释的变异。
#install.packages("ggplot2")
#install.packages("dplyr")
#install.packages("plyr")
library(ggplot2)
library(dplyr)
library(plyr)
pca.rotation <- pca.results$rotation
pca.rotation
pca.pv <- summary(pca.results)$importance[2,]
pca.pv
# PC1 PC2 PC3 PC4 PC5
# 0.66986 0.20273 0.06785 0.03502 0.02454
调整数据结构,用于作图
提取前两个主成分构建数据框,并增加分组信息列
low_dim_df <- as.data.frame(pca.results$x[,c(1,2)])
low_dim_df$group <- meta_df$group
#查看前3行
low_dim_df[1:3,]
下面以PCA分析得到的"low_dim_df"作为输入,画图
画置信区间圆圈的函数
先运行函数 add_ellipase,用来增加置信区间椭圆线。
通过调整参数,以达到你想要的效果,参数含义为:
- p: ggplot2返回的对象
- x,y: 主成分的列名
- group: 分组列
- ellipse_pro: 置信区间,默认0.95
- linetype可选类型: blank, solid, dashed, dotted, dotdash, longdash, twodash
- colour:颜色
- lwd:线的粗细
add_ellipase <- function(p, x="PC1", y="PC2", group="group",
ellipase_pro = 0.95,
linetype="dashed",
colour = "black",
lwd = 2,...){
obs <- p$data[,c(x, y, group)]
colnames(obs) <- c("x", "y", "group")
ellipse_pro <- ellipase_pro
theta <- c(seq(-pi, pi, length = 50), seq(pi, -pi, length = 50))
circle <- cbind(cos(theta), sin(theta))
ell <- ddply(obs, 'group', function(x) {
if(nrow(x) <= 2) {
return(NULL)
}
sigma <- var(cbind(x$x, x$y))
mu <- c(mean(x$x), mean(x$y))
ed <- sqrt(qchisq(ellipse_pro, df = 2))
data.frame(sweep(circle %*% chol(sigma) * ed, 2, mu, FUN = '+'))
})
names(ell)[2:3] <- c('x', 'y')
ell <- ddply(ell, .(group) , function(x) x[chull(x$x, x$y), ])
p <- p + geom_polygon(data = ell, aes(x=x,y=y,group = group),
colour = colour,
alpha = 1,fill = NA,
linetype=linetype,
lwd =lwd)
return(p)
}
 发表于 2025-5-31 08:39:21
发表于 2025-5-31 08:39:21
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )