背景
模仿paper里方法做GSVA,结果保存到文件,能作为输入文件画出bar plot。

出自文章PMID: 29988129
应用场景
关于GSVA分析,可以这样理解:如果说表达矩阵反映了样本和基因的关系,则GSVA将一个“样本×基因”的矩阵转化为“样本×通路”的矩阵,可以反应样本和通路之间的联系。既然用limma包做差异表达分析可以寻找样本间差异表达的基因,那么,使用limma包对GSVA的结果(依然是一个矩阵)做同样的分析,则可以寻找样本间有显著差异的通路。这些“差异表达”的通路,相对于基因而言,更加具有生物学意义,更具有可解释性,是统计学与生物学成功结合后,对GSEA结果的一次升华,可以进一步用于肿瘤subtype的分型等等与生物学意义结合密切的探究。
示例文件
not_easy_input_expr.csv是基因表达数据。
- 此处gsva_limma.csv文件是用通路做差异分析获得的。
环境设置
安装包
#使用国内镜像安装包
options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
install.packages("msigdbr")
加载包
library(msigdbr)
library(dplyr)
library(data.table)
library(GSVA)
library(limma)
library(stringr)
library(ggplot2)
Sys.setenv(LANGUAGE = "en") #显示英文报错信息
options(stringsAsFactors = FALSE) #禁止chr转成factor
# options(mc.cores = parallel::detectCores())
输入文件
GSVA需要两个输入文件:基因表达矩阵和通路里的基因列表。
- hallmark.gs.RData:“整理通路里的基因列表”这部分生成的,同一物种同一版本注释可重复使用。
- easy_input_expr.csv:基因表达矩阵,每行一个基因,每列一个sample。
整理通路里的基因列表
下面这部分运行后,要保存好生成的hallmark.gs.RData文件,同一物种同一版本注释可重复使用。以后就可以跳过这步,直接进入“GSVA”。
每次只需要更换表达矩阵,用同一物种同一版本gmt生成的"hallmark.gs.RData"可以一直用。
为了减少冗余信息的干扰,paper中要求:
- 在每个geneset中,不应该出现重复的基因;
- 在两个或更多个pathway中出现的基因应该被彻底剔除。
在实际分析过程中,笔者留意到在单个的geneset中已经很少出现重复的基因,但在两个或更多个pathway中出现的基因则数目相对较多,读者可酌情考虑,调整此处过滤的力度。毕竟是对通路基因集(分析前提)产生实际影响。
#查看msigdbr包里自带的物种
msigdbr_species()
#其他物种可自行到GSEA官网下载,http://software.broadinstitute.org/gsea/downloads.jsp
h <- msigdbr(species = "Homo sapiens", # 物种拉丁名
category = "H") #此处以hallmark为例,你也可以选择MSigDB的其他注释
# 示例数据表达矩阵的基因名是gene symbol,这里就选gene_symbol。
# 如果你的表达矩阵以ENTREZ ID作为基因名,就把下面这段的gene_symbol换成entrez_gene
h <- select(h, gs_name, gene_symbol) %>% #或entrez_gene
as.data.frame %>%
split(., .$gs_name) %>%
lapply(., function(x)(x$gene_symbol)) #或entrez_gene
# 在每个geneset里面去掉重复的基因
gs <- lapply(h, unique)
# 接下来去掉那些在两个或更多个pathways里出现过的genes
count <- table(unlist(gs))
keep <- names(which(table(unlist(gs)) < 2))
gs <- lapply(gs, function(x) intersect(keep, x))
# 过滤之后,很多pathway一个gene都不剩了,去掉这些
gs <- gs[lapply(gs, length) > 0]
# 预览过滤后的结果
head(gs)
# 保存到文件,方便以后重复使用
save(gs, file = "hallmark.gs.RData")
GSVA
# 读入前面生成的通路中的基因列表
(load("hallmark.gs.RData")) #保存在当前文件夹
# 读入基因表达矩阵
gsym.expr <- read.csv("easy_input_expr.csv", row.names = 1)
head(gsym.expr)
# 首先创建参数对象
gsva_par <- gsvaParam(exprData = as.matrix(gsym.expr),
geneSets = gs)
# 然后进行计算
gsva_es <- gsva(gsva_par)
# 预览GSVA分析返回的矩阵
head(gsva_es)
# 把通路的表达量保存到文件
write.csv(gsva_es, "gsva_output.csv", quote = F)
到这里,GSVA分析就结束了。
接下来,你可能要对这些通路进行进一步的分析,此处以差异表达分析为例。
通路的差异表达分析
# 分组
group_list <- data.frame(sample = colnames(gsva_es), group = c(rep("a", 3), rep("b", 3)))
head(group_list)
# 设置对比
design <- model.matrix(~ 0 + factor(group_list$group))
colnames(design) <- levels(factor(group_list$group))
rownames(design) <- colnames(gsva_es)
design
# 构建差异比较矩阵
contrast.matrix <- makeContrasts(b-a, levels = design)
# 差异分析,b vs. a
fit <- lmFit(gsva_es, design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
x <- topTable(fit2, coef = 1, n = Inf, adjust.method = "BH", sort.by = "P")
head(x)
#把通路的limma分析结果保存到文件
write.csv(x, "gsva_limma.csv", quote = F)
#输出t值,用做barplot的输入数据
pathway <- str_replace(row.names(x), "HALLMARK_", "")
df <- data.frame(ID = pathway, score = x$t)
write.csv(df, "easy_input2_for39bar.csv", quote = F, row.names = F)
开始画图
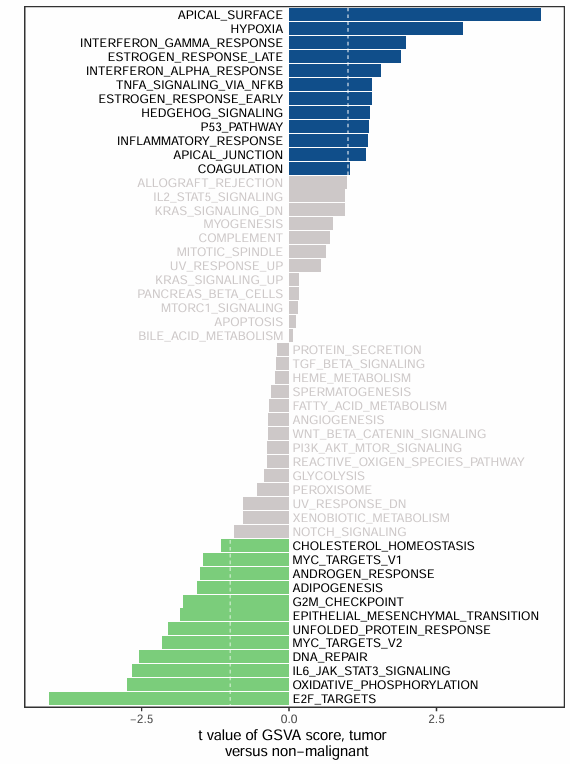
t为柱子的长度(图中横轴),行名为图中pathway的名称。
如果想做更多细节上的调整,可参考FigureYa39bar。
df <- read.csv("easy_input2_for39bar.csv")
head(df)
#按照score的值分组
cutoff <- 1
df$group <- cut(df$score, breaks = c(-Inf, -cutoff, cutoff, Inf),labels = c(1,2,3))
#按照score排序
sortdf <- df[order(df$score),]
sortdf$ID <- factor(sortdf$ID, levels = sortdf$ID)
head(sortdf)
ggplot(sortdf, aes(ID, score, fill = group)) + geom_bar(stat = 'identity') +
coord_flip() +
scale_fill_manual(values = c('palegreen3', 'snow3', 'dodgerblue4'), guide = FALSE) +
#画2条虚线
geom_hline(yintercept = c(-cutoff,cutoff),
color="white",
linetype = 2, #画虚线
size = 0.3) + #线的粗细
#写label
geom_text(data = subset(df, score < 0),
aes(x=ID, y= 0, label= paste0(" ", ID), color = group),#bar跟坐标轴间留出间隙
size = 3, #字的大小
hjust = "inward" ) + #字的对齐方式
geom_text(data = subset(df, score > 0),
aes(x=ID, y= -0.1, label=ID, color = group),
size = 3, hjust = "outward") +
scale_colour_manual(values = c("black","snow3","black"), guide = FALSE) +
xlab("") +ylab("t value of GSVA score, tumor \n versus non-malignant")+
theme_bw() + #去除背景色
theme(panel.grid =element_blank()) + #去除网格线
theme(panel.border = element_rect(size = 0.6)) + #边框粗细
theme(axis.line.y = element_blank(), axis.ticks.y = element_blank(), axis.text.y = element_blank()) #去除y轴
ggsave("gsva.pdf", width = 6, height = 8)
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )

 发表于 2025-9-8 09:51:28
发表于 2025-9-8 09:51:28
