背景
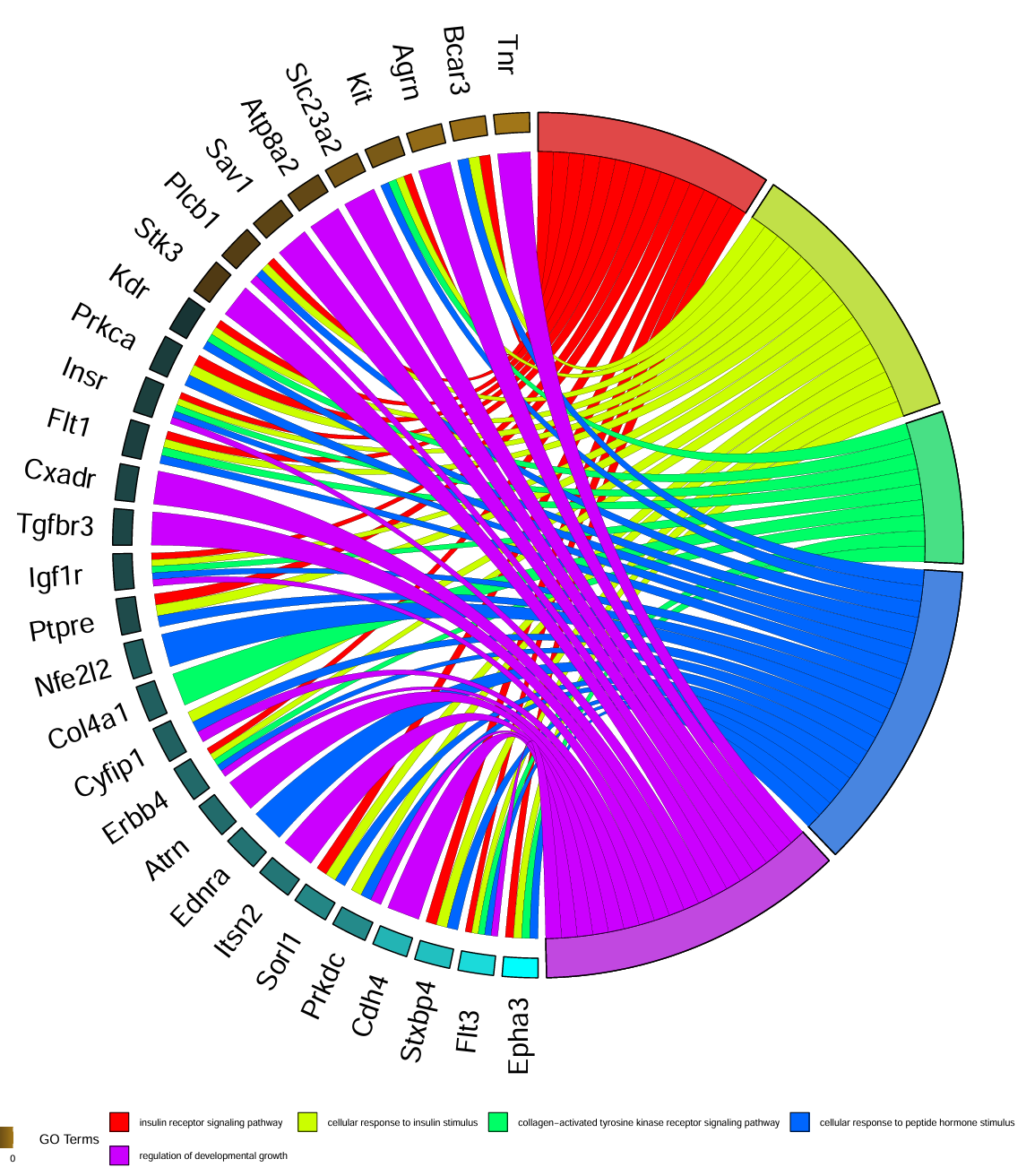
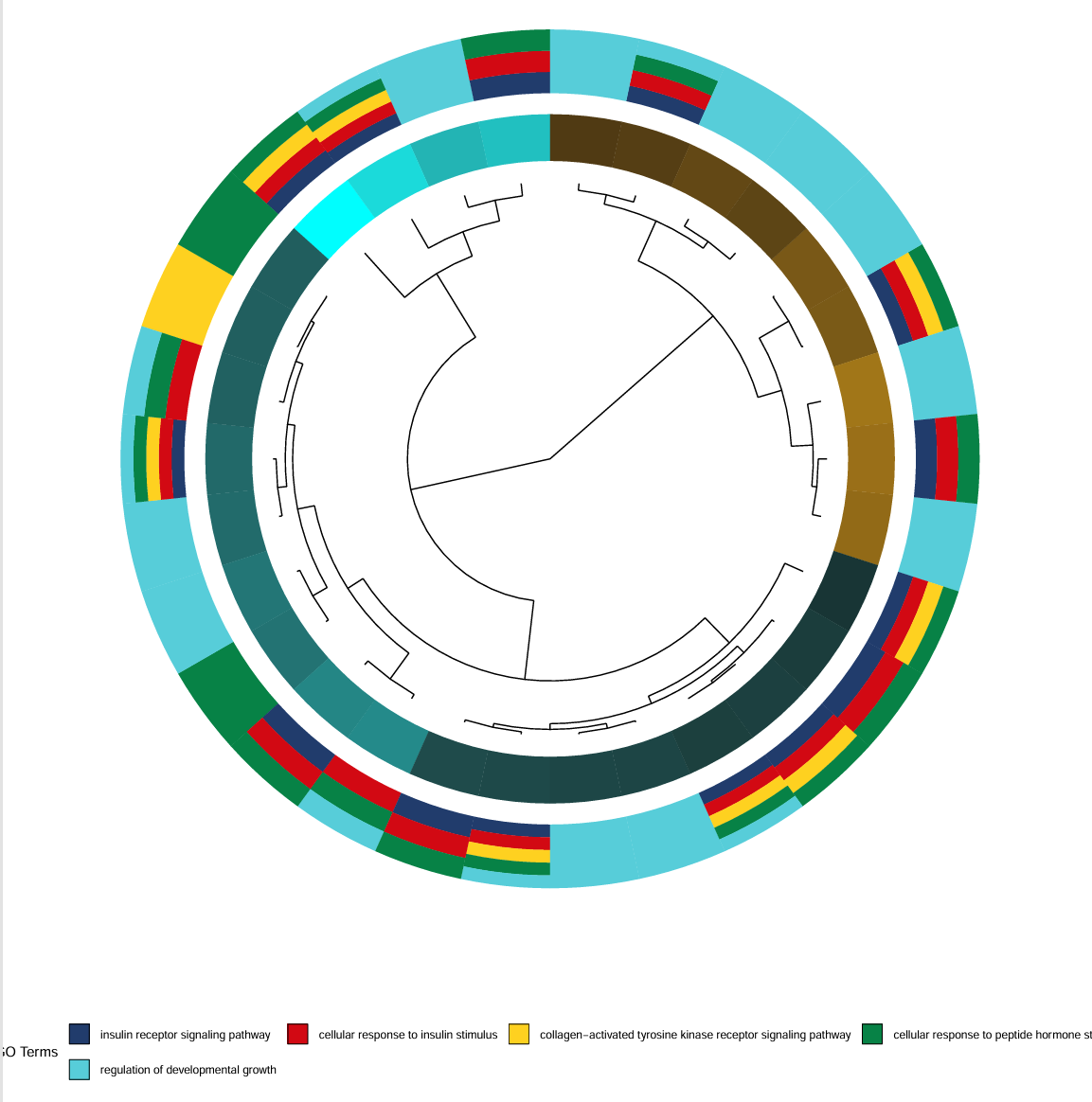
用GOplot展示clusterProfiler的富集分析结果,画出paper里的⭕️图
出自文章PMID: 29066820
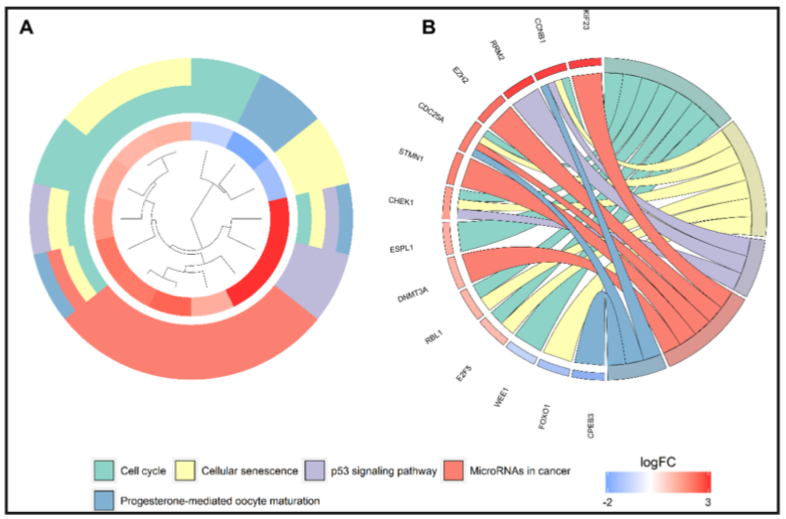
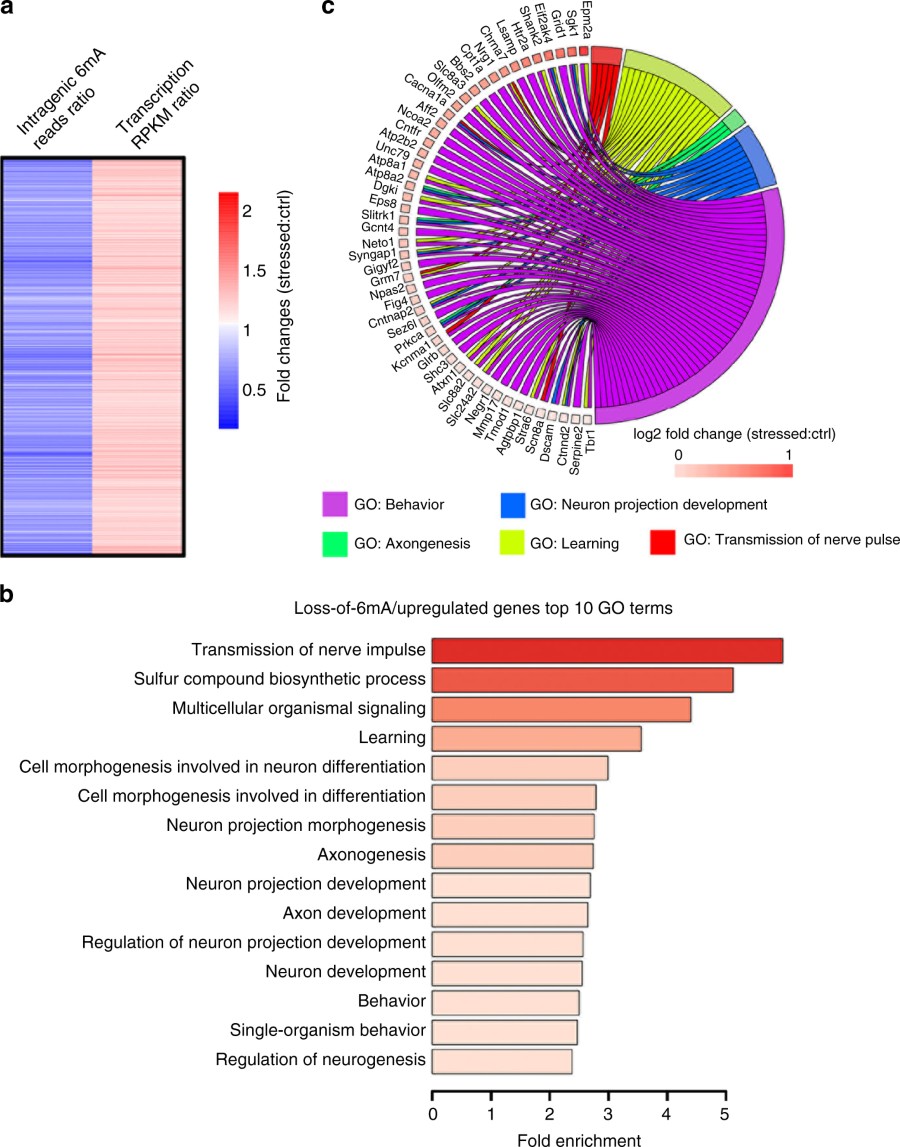
出自文章PMID: 30092571
应用场景
适用于:既想用clusterProfiler做富集分析,又想用GOplot展示结果,但是不知道二者怎样衔接的小伙伴。
此处以GO为例,获得clusterProfiler的富集分析结果,生成GOplot所需的格式,用GOplot画⭕️图。
clusterProfiler除了擅长做GO富集分析以外,还可以用KEGG、Diseaes、Reactome、DAVID、MSigDB等注释库做富集分析。另外,enrichplot自带的gseaplot2函数可以生成GSEA结果的矢量图、多条pathway画到同一个图上,完美代替Java版本的GSEA。
环境设置
安装需要的包
#使用国内镜像安装包
options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler", version = "3.8")
BiocManager::install("org.Mm.eg.db", version = "3.8")
install.packages('GOplot')
加载需要用到的包
library(clusterProfiler)
library(AnnotationHub)
library(AnnotationDbi)
library(GOplot)
library(ggplot2)
Sys.setenv(LANGUAGE = "en") #显示英文报错信息
options(stringsAsFactors = FALSE) #禁止chr转成factor
获取ENTREZ ID跟基因名的对应关系
如果你的基因ID已经是ENTREZ ID,并保存成 very_easy_input_**.csv的格式,就可以跳过这步,直接进入“富集分析”
根据基因名gene symbol找到相应的ensembl ID。模式生物和非模式生物的转换代码稍有不同,根据自己的物种,选择运行以下两段之一。
人和模式生物ENTREZ ID的获取
此处以例文中的小鼠数据为例,其他18个物种只需更改 OrgDb = "org.Mm.eg.db"参数
具体物种对应的R包名字看这页:http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
如果你关心的物种不在这个列表里,请跳过这步,直接进入“非模式生物ENTREZ ID的获取”
gsym.fc <- read.csv("easy_input_Mm.csv", as.is = T)
dim(gsym.fc) # 201
#查看有哪些ID
#keytypes(org.Mm.eg.db)
gsym.id <- bitr(gsym.fc$SYMBOL, #基因名
fromType = "SYMBOL", #从gene symbol
toType = "ENTREZID", #提取ENTREZ ID
OrgDb = "org.Mm.eg.db") #相应物种的包,人是org.Hs.eg.db
head(gsym.id) # 197
#合并基因名、ENTREZID、foldchange
idvec <- gsym.id$ENTREZID
names(idvec) <- gsym.id$SYMBOL
gsym.fc$ENTREZID <- idvec[gsym.fc$SYMBOL]
head(gsym.fc) # 没有的就是空值
#保存到文件
write.csv(gsym.fc[,c(3,2)], "very_easy_input_Mm.csv", quote = F, row.names = F)
非模式生物ENTREZ ID的获取
模式生物请跳过这段,直接进入“富集分析”。
准备工作:
把基因名跟ENTREZ ID对应关系的注释文件保存到本地文件,以后想要获取同一基因组版本的ENTREZ ID时,直接导入这个“Zmays(物种名).AH66225(版本).sqlite”文件就可以了。此处以玉米为例:
hub <- AnnotationHub() #大概需要2分钟,网速慢就要更久
#查看AnnotationHub里有哪些物种,记住idx列里的AH***
#d <- display(hub)
#或者直接搜zea(玉米拉丁名的一部分)
# query(hub, "zea") #3.8 biocversion
query(hub,'org.Zea_mays.eg.sqlite') # 3.21 biocversion
#此处下载“AH61838” AH119718 新版号
maize.db <- hub[['AH119718']] #大概需要3分钟,网速慢就要更久
# BiocManager::version()
#查看包含的基因数
length(keys(maize.db)) #49729
#查看包含多少种ID
columns(maize.db)
#查看前几个基因的ID长啥样
select(maize.db, keys(maize.db)[1:3],
c("REFSEQ", "SYMBOL"), #你想获取的ID
"ENTREZID")
#保存到文件
saveDb(maize.db, "Zmays.AH119718.sqlite")
根据基因名gene symbol获取ENTREZ ID
#读入差异基因
gsym.fc <- read.csv("easy_input_Zm.csv")
head(gsym.fc)
dim(gsym.fc) # 1000
#读入前面保存到本地的注释文件
maize.db <- loadDb("Zmays.AH119718.sqlite")
gsym.id <- bitr(gsym.fc$SYMBOL, #基因名所在的列
"SYMBOL", #根据gene symbol
"ENTREZID", #提取ENTREZ ID
maize.db) #玉米注释
head(gsym.id)
dim(gsym.id) # 190
#合并基因名、ENTREZID、foldchange
idvec <- gsym.id$ENTREZID
names(idvec) <- gsym.id$SYMBOL
gsym.fc$ENTREZID <- idvec[gsym.fc$SYMBOL]
head(gsym.fc)
dim(gsym.fc)
#保存到文件
write.csv(gsym.fc[,c(3,2)], "very_easy_input_Zm.csv", quote = F, row.names = F)
富集分析
参考资料:http://bioconductor.org/packages/release/bioc/vignettes/clusterProfiler/inst/doc/clusterProfiler.html#supported-organisms
如果你已经做好了富集分析,并且保存成“enrichGO_output.csv”的格式,就可以跳过这部分,直接进入“把clusterProfiler输出的富集分析结果转成GOplot所需的格式”
#ENTREZ ID位于第一列,log2foldchange位于第二列
id.fc <- read.csv("very_easy_input_Mm.csv", as.is = T)
head(id.fc)
dim(id.fc)
ego <- enrichGO(gene = id.fc$ENTREZID,
#小鼠用这行
OrgDb = org.Mm.eg.db,
#人类用这行
#OrgDb = org.Hs.eg.db,
#非模式生物用这行,例如玉米
#OrgDb = maize.db,
ont = "BP", #或MF或CC
pAdjustMethod = "BH",
#pvalueCutoff = 0.001,
qvalueCutoff = 0.01)
dim(ego)
#把富集分析结果保存到文件
write.csv(ego,"enrichGO_output.csv",quote = F)
富集的GO term有些是相似的,可以用语义学方法,合并相似的GO term。需要较长时间。
参考资料:https://guangchuangyu.github.io/2015/10/use-simplify-to-remove-redundancy-of-enriched-go-terms/
ego2 <- simplify(ego, cutoff = 0.7, by = "p.adjust", select_fun = min)
dim(ego2)#178
write.csv(ego2,"enrichGO_simplify_output.csv",quote = F)
用enrichplot自带的函数画⭕️
参考资料:https://yulab-smu.top/biomedical-knowledge-mining-book/enrichplot.html
第15项
#把ENTREZ ID转为gene symbol
egox <- setReadable(ego, 'org.Mm.eg.db', #物种
'ENTREZID')
#把基因倍数信息转成画图所需的格式
geneList <- id.fc$log2fc
names(geneList) <- id.fc$ENTREZID
#画⭕️图
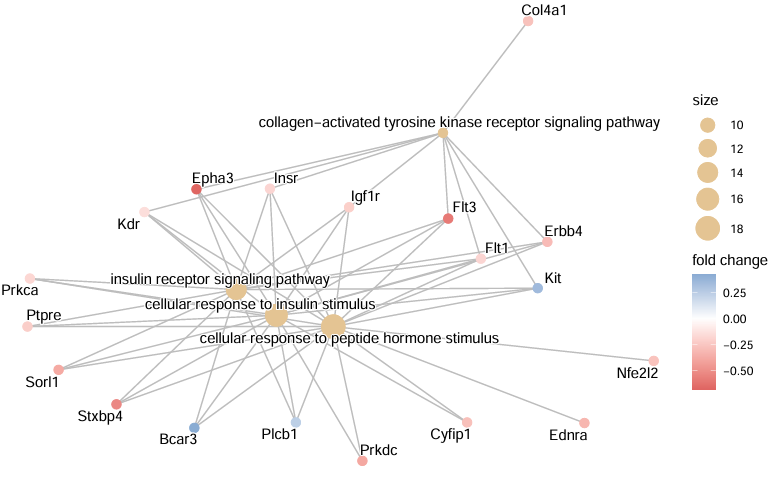
cnetplot(egox,
foldChange = geneList,
#foldChange = NULL, #不展示倍数
circular = TRUE,
#node_label = FALSE, #如果太多,就不要显示基因名了
showCategory = 4, #显示富集的term数量,默认5
colorEdge = TRUE)
#保存到pdf文件
ggsave("clusterProfiler_circle.pdf", width = 8, height = 5)
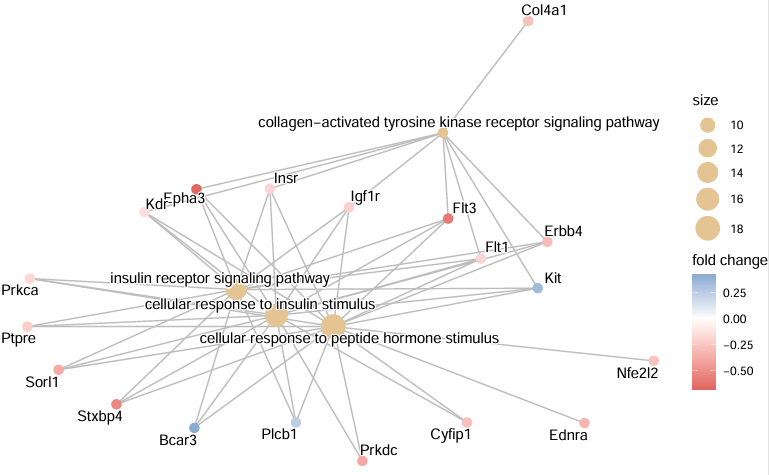
#不画成⭕️,process分开,效果更好呢
cnetplot(egox,
foldChange = geneList,
#foldChange = NULL, #不展示倍数
#circular = TRUE,
#node_label = FALSE, #不显示基因名
showCategory = 4, #显示的富集term数量,默认5
colorEdge = TRUE)
ggsave("clusterProfiler_not_circle.pdf", width = 8, height = 5)
 发表于 2025-9-7 20:14:07
发表于 2025-9-7 20:14:07
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )