背景
在转录组学研究领域,差异表达分析是揭示基因功能与调控机制的核心技术手段。当我们完成RNA测序(RNA-Seq)实验后,首先获得的是原始的读取计数(count)数据——这些看似简单的数字矩阵,却蕴含着解读基因表达调控奥秘的关键信息。尽管存在FPKM、TPM等标准化表达量指标,但主流差异分析工具如DESeq2和edgeR都明确要求输入原始count数据。这看似违背直觉的选择背后,实则蕴含着深刻的统计学原理和生物学意义。
1. 差异表达分析的本质与挑战
差异表达分析的核心目标,是从海量基因中识别出在不同实验条件下(如疾病vs健康、处理vs对照)表达水平发生显著变化的基因。这一过程面临两个主要技术挑战:
• 技术偏差的干扰:包括测序深度(样本间测序总量差异)、基因长度(长基因更易被测到)、GC含量偏好等非生物因素
• 数据分布特性的保持:基因表达数据具有独特的统计分布特征,需选择适配的分析模型
在早期RNA-Seq分析方法中,研究人员常借鉴微阵列芯片的分析思路,使用FPKM(Fragments Per Kilobase per Million)或TPM(Transcripts Per Million)等标准化数据,结合t检验、ANOVA等参数检验方法。然而,随着对RNA-Seq数据特性的深入理解,这种看似合理的方式被发现存在根本性缺陷。
2. 原始Count数据的本质特性
2.1 什么是Count数据?
原始count数据本质上是比对到每个基因的测序reads(或fragments)数量。例如,某基因在样本A中检测到100个reads,在样本B中检测到50个reads,直观反映了该基因在A中的表达量是B中的两倍。
数学上,RNA-Seq的count数据具有以下关键特性:
• 离散性:取值只能是非负整数(0,1,2,...)
• 高方差性:表达量均值与方差存在特定关系(方差通常大于均值)
• 依赖测序深度:总测序深度越大,各基因的count预期值越高
2.2 统计分布特征
Count数据天然符合离散型概率分布,尤其是负二项分布(Negative Binomial Distribution)。这一分布有两个关键参数:
- 均值μ:基因在特定条件下的平均表达水平
- 离散度α:描述方差与均值的关系(方差= μ + αμ²)
负二项分布能够精确捕捉RNA-Seq数据中普遍存在的过度离散(over-dispersion)现象——即基因表达方差大于其均值的特性。这一特性源于生物学和技术性变异的共同作用。
3. 为何Count数据在差异分析中更受青睐?
3.1 统计模型的完美适配
DESeq2和edgeR等现代差异分析工具的核心算法是围绕count数据的负二项分布特性设计的:
-
精确的方差建模:
方差 = μ + αμ²
其中μ为基因表达均值,α为离散度参数。这种建模能准确捕获基因表达变异性随均值增加而增加的特性。
-
信息“借用”策略:
• 利用所有基因的数据估计全局离散度趋势
• 对低表达基因“借用”高表达基因的变异信息,提高统计检验的稳健性。
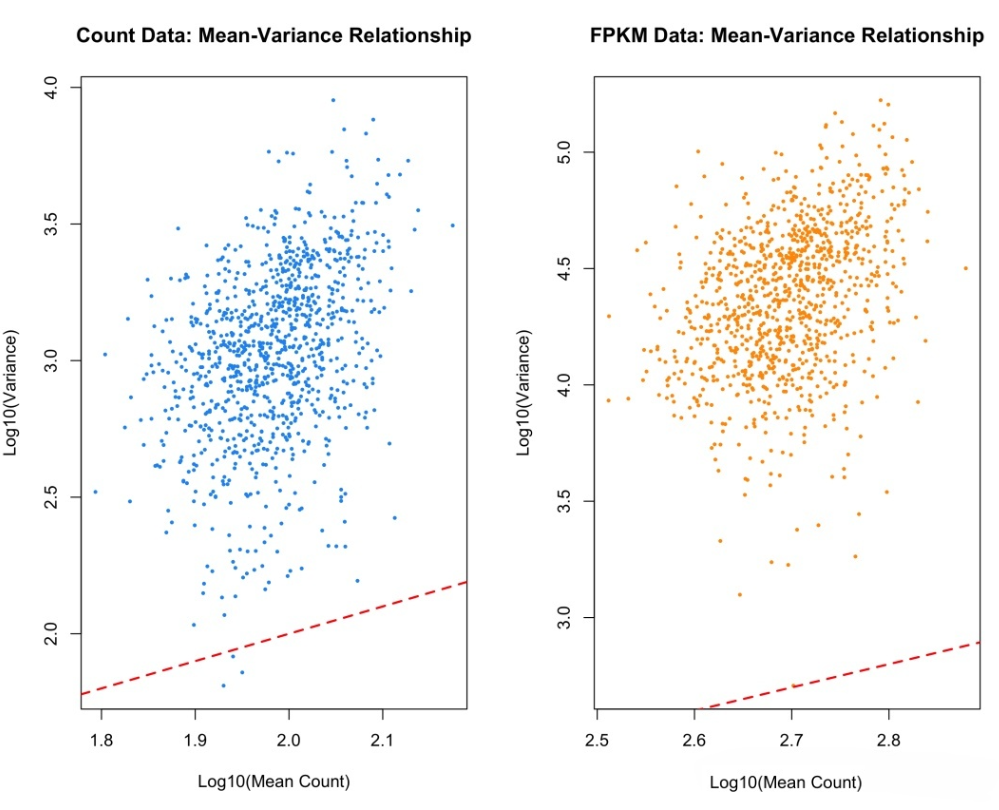
Count数据与FPKM数据的均值-方差关系对比。Count数据展现出清晰的二次趋势,而FPKM数据则呈现不规则分布。
相比之下,FPKM/TPM等标准化数据破坏了原始数据的离散特性,将其转换为连续变量。这种转换导致:
• 数据不再符合负二项分布
• 方差-均值关系被扭曲
• 标准参数检验(如t检验)的基本假设被违反
• 尤其对低表达基因的分析准确性大幅降低
3.2 信息保留与低表达基因的敏感性
FPKM/TPM标准化在消除技术偏差的同时,也引入了信息损失,尤其对低表达基因影响显著:
• “压缩效应”:低count值经标准化后趋向于接近零的小数值
• 分辨率丧失:例如,原始count从1增加到2(翻倍变化)可能被标准化为0.01到0.02,在后续分析中被忽略
• 假阴性风险:真正的低表达差异基因易被遗漏
原始count数据则完整保留了低丰度转录本的表达变化信息。结合DESeq2/edgeR的统计模型,即使是微弱的生物学信号也能被有效捕捉。
4. 主流工具的内部标准化机制
4.1 内置的稳健标准化
使用原始count数据不等于忽视标准化需求。DESeq2和edgeR在分析流程中内置了更精细的标准化步骤:
• DESeq2的median-of-ratios方法:
s_j = median_{i} ( \frac{g_{ij}}{ \prod_{k=1}^m g_{ik}^{1/m} } )
其中gᵢⱼ为基因i在样本j中的count值。该方法使用几何平均计算每个基因的参考值,再取中位数确定样本特异的尺度因子。
• edgeR的TMM(Trimmed Mean of M-values)方法:
基于大部分基因未差异表达的假设,筛选表达丰度适中且倍数变化较小的基因子集计算标准化因子。
这些方法专门针对count数据设计,能更稳健地处理样本间的组成差异和离群值影响。
4.2 工作流程对比
基于Count数据的差异分析工作流程与传统流程对比
| 步骤 |
Count+DESeq2/edgeR流程 |
FPKM/TPM+传统检验流程 |
| 数据输入 |
原始count矩阵 |
标准化表达矩阵 |
| 标准化方式 |
基于负二项分布模型的内置标准化 |
外部标准化 |
| 统计检验 |
基于离散性数据的Wald检验或LRT |
t检验、ANOVA等 |
| 低表达处理 |
借用信息策略提高敏感性 |
易被过滤或忽略 |
| 结果可靠性 |
高(模型与数据匹配) |
中低(模型假设不符) |
5. 实践建议与常见误区
5.1 数据分析操作指南
-
数据准备阶段:
• 保留原始count矩阵(基因×样本)
• 避免预先转换为FPKM/TPM等标准化值
• 使用质量控制工具(如FastQC、MultiQC)评估原始数据质量
-
分析工具选择:
# DESeq2标准分析流程
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = count_data,
colData = sample_info,
design = ~ group)
dds <- DESeq(dds)
res <- results(dds)
-
过滤策略:
• 在工具内部执行低表达过滤(非强制)
• 推荐方法:去除在所有样本中count总和过低的基因
# 在DESeq2中自动过滤
dds <- dds[rowSums(counts(dds)) >= 10, ]
5.2 避免常见误区
- 误区一:“标准化数据更准确,应替代count数据”
• 纠正:FPKM/TPM适用于样本间基因表达比较和可视化,但差异分析必须使用count数据
- 误区二:“低表达基因应全部过滤”
• 纠正:DESeq2/edgeR能有效利用低表达基因信息,过早过滤可能导致关键生物信号丢失
- 误区三:“不同工具的结果应完全一致”
• 纠正:DESeq2和edgeR算法存在差异,结果可能有5-10%不一致性,可通过交叉验证提高可靠性
6. 总结:尊重数据的本质特性
在差异表达分析中,原始count数据之所以成为金标准,本质上是尊重数据的统计本质和生物学生成过程。RNA-Seq技术产生的计数数据天然符合离散分布特性,而DESeq2、edgeR等工具专门为此设计,能更准确地捕捉基因表达的生物学差异,尤其在处理低表达基因和样本间变异时优势显著。
理解这一原理不仅有助于研究者正确选择分析方法,更体现了生物信息学中一个普适原则:数据分析方法应适配数据的本质特性,而非强行套用通用模型。在追求精准生物学发现的旅程中,统计严谨性始终是保障结果可靠性的第一道防线。
“在数据科学中,最强大的洞察往往来自于尊重数据的本质特性,而非强行将其塞入预设的模型框架。” ——生物信息学箴言
随着单细胞转录组、空间转录组等新技术的发展,count数据的分析方法仍在持续演进。但对数据生成原理的深刻理解与尊重,始终是解锁生物学奥秘的金钥匙。
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )

 发表于 2025-8-4 12:22:30
发表于 2025-8-4 12:22:30