[i=s] 本帖最后由 生信喵 于 2025-6-4 10:10 编辑 [/i]
背景
前面我们下载了新版tcga的rnaseq counts数据。
接下来就利用TCGA数据库的LIHC数据进行数据整理,获得我们进行差异分析需要的counts、TPM等数据。
一、处理代码
### 解压数据,创建存储文件夹----
# 我们使用命令行下载的,直接就是解压后的文件目录,可以跳过这一步
# setwd("TCGA-LIHC") # 设置工作路径
# dir.create('RawMatrix/') # 新建文件夹存储下载的原始数据
# tar_file <- "./gdc_download_20241222_135942.082516.tar.gz"
# extract_dir <- "./RawMatrix"
# untar(tar_file, exdir = extract_dir) # 导入tar.gz,并解压文件
#数据准备----
# C:/Users/Administrator/project_gdc2/RawMatrix 是数据目录
rm(list = ls()) #### 魔幻操作,一键清空~
getwd()
setwd('C:/Users/Administrator/project_gdc2')
dir.create('RawData/') # 新建文件夹存储count/TPM/差异表达矩阵等txt格式
dir.create('RawData/csv/') # 新建文件夹存储csv格式的矩阵
### 数据整理----
library(data.table)
library(dplyr)
sample_sheet <- fread("./gdc_sample_sheet.2024-12-22.tsv") # 读取样本信息
sample_sheet$Barcode <- substr(sample_sheet$`Sample ID`,1,15) # 取ID前15字符作为barcode
sample_sheet1 <- sample_sheet %>% filter(!duplicated(sample_sheet$Barcode)) # 去重
sample_sheet2 <- sample_sheet1 %>% filter(grepl("01$|11$|06$",sample_sheet1$Barcode))
# table(as.numeric(substr(sample_sheet1$Barcode,14,15) < 10) == 1)
# 0 1
# 50 373
# sample_sheet1$Barcode[!grepl("01$|11$|06$",sample_sheet1$Barcode)]
# [1] "TCGA-DD-AACA-02" "TCGA-ZS-A9CF-02"
# 02是可以用的,也是肿瘤样本。直接用sample_sheet1。
# 发现02的同时也有01样本,所以是要移除后使用。
# Barcode的最后两位:01表示肿瘤样本,11表示正常样本,06表示转移样本
# A:Vial, 在一系列患者组织中的顺序,绝大多数样本该位置编码都是A; 很少数的是B,表示福尔马林固定石蜡包埋组织,已被证明用于测序分析的效果不佳,所以不建议使用-01B的样本数据:
# 02也是要的
TCGA_LIHC_Exp <- fread("./RawMatrix/0036fcec-eaed-430b-9a23-5efb2d2cc7f2/32b682ec-8156-44ca-bff0-26155c7fdc12.rna_seq.augmented_star_gene_counts.tsv") # 任意读取一个文件
# 创建包含"gene_id","gene_name","gene_type"的数据框,用于合并表达数据
TCGA_LIHC_Exp <- TCGA_LIHC_Exp[-c(1:4),c("gene_id","gene_name","gene_type")]
### 将所有样本合并成一个数据框
for (i in 1:nrow(sample_sheet2)) {
folder_name <- sample_sheet2$`File ID`[i]
file_name <- sample_sheet2$`File Name`[i]
sample_name <- sample_sheet2$Barcode[i]
data1 <- fread(paste0("./RawMatrix/",folder_name,"/",file_name))
#unstranded代表count值;如果要保存TPM,则改为 tpm_unstranded
data2 <- data1[-c(1:4),c("gene_id","gene_name","gene_type","tpm_unstranded")]
colnames(data2)[4] <- sample_name
TCGA_LIHC_Exp <- inner_join(TCGA_LIHC_Exp,data2)
}
### 根据需要的表达比例筛选满足条件的基因
zero_percentage <- rowMeans(TCGA_LIHC_Exp[, 4:ncol(TCGA_LIHC_Exp)] == 0)
TCGA_LIHC_Exp1 <- TCGA_LIHC_Exp[zero_percentage < 0.6, ] # 筛选出超过60%样本中存在表达的基因
#28842
dim(TCGA_LIHC_Exp1)
TCGA_LIHC_Exp1 = avereps(TCGA_LIHC_Exp1[,-c(1:3)],ID = TCGA_LIHC_Exp1$gene_name) # 对重复基因名取平均表达量,并将基因名作为行名
dim(TCGA_LIHC_Exp1)# 28776 421
# tpm到这里就可以,不用筛选低表达
### 创建样本分组
library(stringr)
tumor <- colnames(TCGA_LIHC_Exp1)[substr(colnames(TCGA_LIHC_Exp1),14,15) == "01"]
normal <- colnames(TCGA_LIHC_Exp1)[substr(colnames(TCGA_LIHC_Exp1),14,15) == "11"]
tumor_sample <- TCGA_LIHC_Exp1[,tumor]
normal_sample <- TCGA_LIHC_Exp1[,normal]
exprSet_by_group <- cbind(tumor_sample,normal_sample)
dim(exprSet_by_group) # 28776 421
gene_name <- rownames(exprSet_by_group)
exprSet <- cbind(gene_name, exprSet_by_group) # 将gene_name列设置为数据框的行名,合并后又添加一列基因名
### 存储TPM数据
fwrite(exprSet,"./RawData/TCGA_LIHC_Tpm.txt") # txt格式
write.csv(exprSet, "./RawData/csv/TCGA_LIHC_Tpm.csv", row.names = FALSE) # csv格式
TCGA_LIHC_Exp1 = avereps(TCGA_LIHC_Exp[,-c(1:3)],ID = TCGA_LIHC_Exp$gene_name) # 对重复基因名取平均表达量,并将基因名作为行名
TCGA_LIHC_Exp1 <- TCGA_LIHC_Exp1[rowMeans(TCGA_LIHC_Exp1)>100,] # 根据需要去除低表达基因,这里设置的平均表达量100为阈值
dim(TCGA_LIHC_Exp1)#13526
### 创建样本分组
library(stringr)
tumor <- colnames(TCGA_LIHC_Exp1)[substr(colnames(TCGA_LIHC_Exp1),14,15) == "01"]
normal <- colnames(TCGA_LIHC_Exp1)[substr(colnames(TCGA_LIHC_Exp1),14,15) == "11"]
tumor_sample <- TCGA_LIHC_Exp1[,tumor]
normal_sample <- TCGA_LIHC_Exp1[,normal]
exprSet_by_group <- cbind(tumor_sample,normal_sample)
dim(exprSet_by_group)
gene_name <- rownames(exprSet_by_group)
exprSet <- cbind(gene_name, exprSet_by_group) # 将gene_name列设置为数据框的行名,合并后又添加一列基因名
### 存储counts和TPM数据
# fwrite(exprSet,"./RawData/TCGA_LIHC_Count.txt") # txt格式
# write.csv(exprSet, "./RawData/csv/TCGA_LIHC_Count.csv", row.names = FALSE) # csv格式
fwrite(exprSet,"./RawData/TCGA_LIHC_Tpm.txt") # txt格式
write.csv(exprSet, "./RawData/csv/TCGA_LIHC_Tpm.csv", row.names = FALSE) # csv格式
二、差异表达分析
这里分享用DeSeq2 R包进行差异分析的代码:
rm(list = ls()) #### 魔幻操作,一键清空~
getwd()
setwd('C:/Users/Administrator/project_gdc2')
library(DESeq2)
library(stringr)
options(datatable.fread.datatable=FALSE)#保证fread返回数据框
exp <- fread('./RawData/TCGA_LIHC_Count.txt')
# exp <- read.table('./RawData/TCGA_LIHC_Count.txt',sep = ',',header = T,row.names = 1)
rownames(exp) <- exp$gene_name
exp <- exp[,-1]
group_list <- ifelse(substr(colnames(exp),14,15) == "01",'tumor','normal')
# table(group_list)
group_list <- as.factor(group_list)
# levels(group_list)
# [1] "normal" "tumor" #设置后面的是肿瘤
colData <- data.frame(row.names = colnames(exp),
condition = group_list) # 列出每个样品是肿瘤样品还是正常样品
dds <- DESeqDataSetFromMatrix(countData = round(exp), #取整数
colData = colData,
design = ~ condition) %>%
DESeq() # 将数据框转为DESeq2的数据集类型,然后用DESeq函数做差异分析
# some values in assay are not integers #取整数避免了这个报错
# In DESeqDataSet(se, design = design, ignoreRank) :
# some variables in design formula are characters, converting to factors
# res <- results(dds)
# res <- as.data.frame(res) #tumor/normal
# 提取结果,两两比较
res <- results(dds,
contrast = c("condition",rev(levels(group_list)))
)
# res <- results(dds) #一样
# 按设置的比较水平提取dds的数据(从DESeq分析中提取结果表,给出样本的基本均值,logFC及其标准误,检验统计量,p值和矫正后的p值)。
# rev表示调换顺序,levels(group_list)是因子水平
DEG <- res[order(res$pvalue),] %>%
as.data.frame() # 将res按照P值从小到大排序,并转换回数据框。order函数获取向量每个元素从小到大排序后的第几位
head(DEG)
# 添加change列标记基因上调下调
logFC_cutoff <- with(DEG,mean(abs(log2FoldChange)) + 2*sd(abs(log2FoldChange)))
# 设置logFC的阈值,可以计算得出,也可以设置固定值,例如2。
# abs函数计算log2FoldChange的绝对值。
# 均数+2倍标准差可以包含log2FoldChange的95%置信区间
# logFC_cutoff
# 打标签:logFC > 2 & FDR < 0.05:上调基因,logFC < -2 & FDR < 0.05:下调基因,其它认为无显著差异
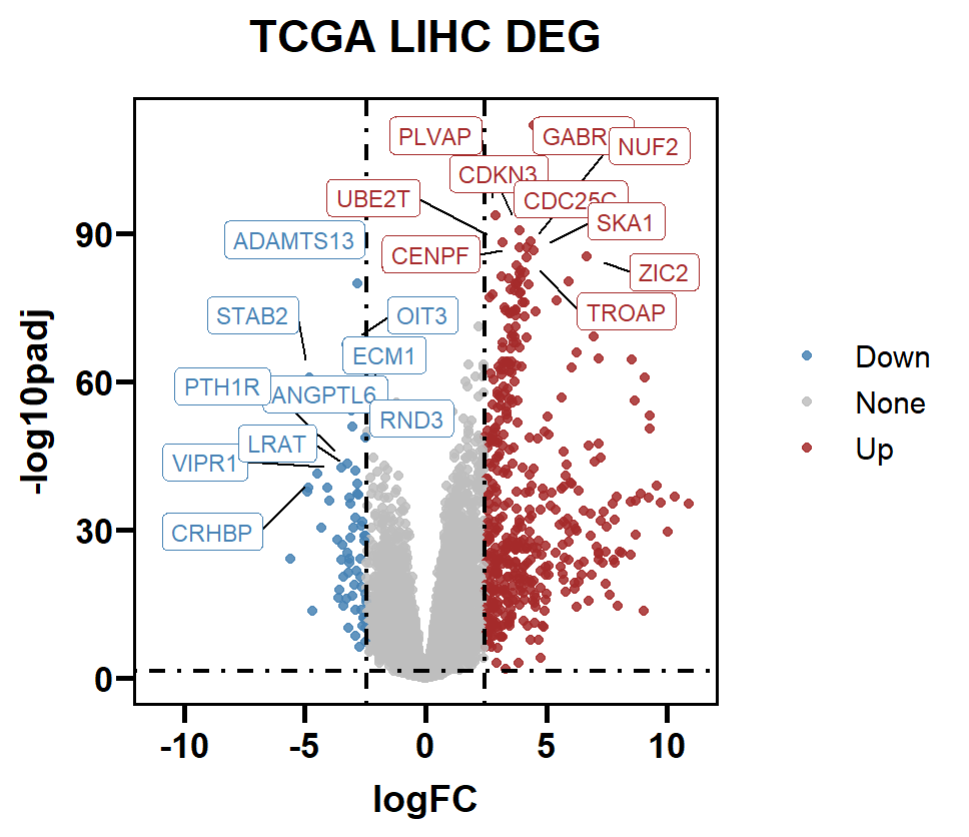
LIHC_DEG <- DEG %>%
mutate(change = case_when(log2FoldChange > logFC_cutoff & padj < 0.05 ~ "Up",
abs(log2FoldChange) < logFC_cutoff | padj > 0.05 ~ "None",
log2FoldChange < -logFC_cutoff & padj < 0.05 ~ "Down"))
table(LIHC_DEG$change)
# Down None Up
# 62 13008 456
# 保存添加标签后的基因
fwrite(LIHC_DEG,"./RawData/02.LIHC_DEG.txt") # txt格式
write.csv(LIHC_DEG, "./RawData/csv/02.LIHC_DEG.csv", row.names = F) # csv格式
 发表于 2024-12-22 22:59:29
发表于 2024-12-22 22:59:29
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )