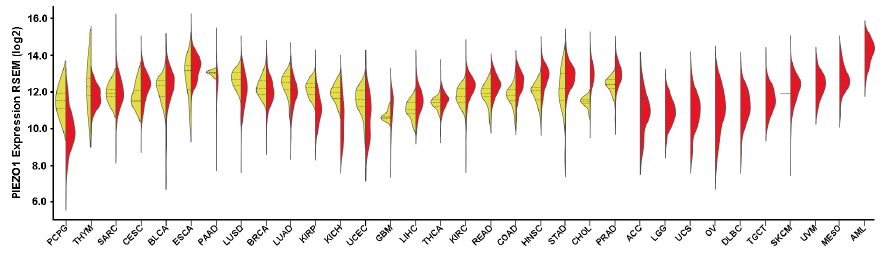
比较现有数据跟GEPIA收录的sample数量
statable<-data.frame()
for (tissuer in sort(unique(tcga_gtex$tissue))){
tcga_gtex_tissuer<-tcga_gtex[tcga_gtex$tissue==tissuer,]
tissuer_tumor_TCGA <- paste0(tissuer,"_tumor_TCGA")
tissuer_normal_TCGA <- paste0(tissuer,"_normal_TCGA")
tissuer_normal_GTEx <- paste0(tissuer,"_normal_GTEx")
tissuerdf<-data.frame(TCGA=tissuer,
TCGA_new_tumor=sum(tcga_gtex_tissuer$type==tissuer_tumor_TCGA),
TCGA_new_normal=sum(tcga_gtex_tissuer$type==tissuer_normal_TCGA),
GTEx_new_num=sum(tcga_gtex_tissuer$type==tissuer_normal_GTEx),
stringsAsFactors=F)
#str(tissuerdf)
statable<-rbind(statable,tissuerdf)
}
cbind(statable, samplepair[,c(1,3:6)])
前4列是现有数据,后面是GEPIA数据
找出每个基因在TPM文件中的行号
idmap <- read.delim("gencode.v23.annotation.gene.probemap",as.is=T)
head(idmap)
system('cut -f 1 gtex_RSEM_gene_tpm >leftgtex') #需要大概1min
tpmid2row <- read.delim("leftgtex",as.is = T)
colnames(tpmid2row) <- "id"
tpmid2row$rownum <- seq(1,nrow(tpmid2row))
head(tpmid2row)
gene2id2row <- merge(idmap,tpmid2row,by="id")
gene2id2row <- gene2id2row[order(gene2id2row$rownum),]
head(gene2id2row)
提取目的基因在肿瘤和正常组织的TPM
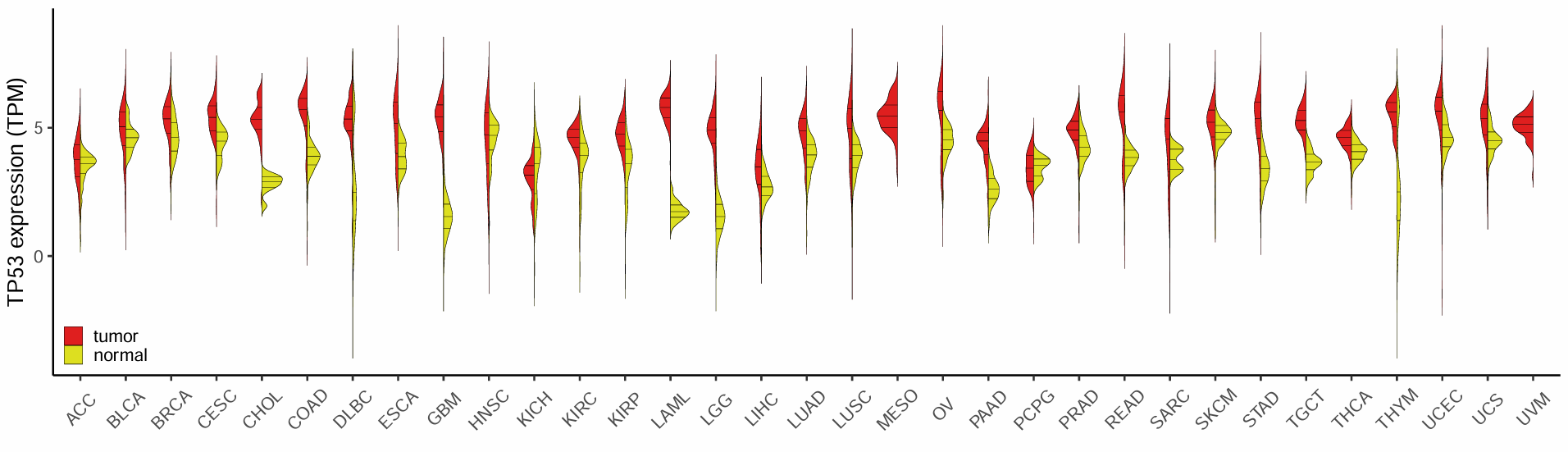
此处以TP53基因为例,提取它的TPM
generownum <- gene2id2row[gene2id2row$gene=="TP53",]$rownum + 1
shellcmd<-paste0("cut -f 2-",ncol(headgtex)," gtex_RSEM_gene_tpm >gtex_tpm")
system(shellcmd) #wait for 3min
system('paste tcga_RSEM_gene_tpm gtex_tpm >tcga_gtex_tpm') #wait for 15min
system('sed \'1q;d\' tcga_gtex_tpm >tpm_header')
gettpmcmd <- paste0("sed \'",generownum,"q;d\' tcga_gtex_tpm >tpm_gene")
system(gettpmcmd)
system('cat tpm_header tpm_gene >genetpm')
tpm <- read.delim("genetpm",as.is = T)
colnames(tpm) <- str_replace_all(colnames(tpm),"[.]","-")
tpm[,1:6]
tpmvec <- as.vector(t(tpm[,2:ncol(tpm)]))
names(tpmvec) <- colnames(tpm)[2:ncol(tpm)]
tcga_gtex$tpm <- tpmvec[tcga_gtex$sample]
#按组织排序
tcga_gtex <- arrange(tcga_gtex,tissue)
write.csv(tcga_gtex[,c(2,4,5)],"easy_input.csv", quote = F)
输入数据预处理
easy_input.csv:第1列tissue是组织(分组),第2列type2是肿瘤/正常组织(左右分组),第3列tpm是TPM值。
有些组织没有normal,需要识别出来单独画。
tcga_gtex <- read.csv("easy_input.csv", row.names = 1, header = T, as.is = F)
head(tcga_gtex)
tumorlist <- unique(tcga_gtex[tcga_gtex$type2=="tumor",]$tissue)
normallist <- unique(tcga_gtex[tcga_gtex$type2=="normal",]$tissue)
withoutNormal <- setdiff(tumorlist, normallist)
MESO和UVM没有normal
tcga_gtex$type2 <- factor(tcga_gtex$type2,levels=c("tumor","normal"))
tcga_gtex_withNormal <- tcga_gtex[!(tcga_gtex$tissue %in% withoutNormal),]
tcga_gtex_MESO <- tcga_gtex[tcga_gtex$tissue=="MESO",]
tcga_gtex_UVM <- tcga_gtex[tcga_gtex$tissue=="UVM",]
开始画图
要用到SplitViolin的图层和函数,保存在GeomSplitViolin.R文件中,位于当前文件夹。
source("GeomSplitViolin.R") #位于当前文件夹
p <- ggplot(tcga_gtex_withNormal, aes(x = tissue, y = tpm, fill = type2)) + #x对应肿瘤的类型,y对应表达量,fill填充对应组织的类型
geom_split_violin(draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = "solid", #周围线的轮廓
color = "black", #周围线颜色
size = 0.2,
na.rm = T,
position ="identity")+ #周围线粗细
ylab("TP53 expression (TPM)") + xlab("") +
ylim(-4,9) +
scale_fill_manual(values = c("#DF2020", "#DDDF21"))+
theme_set(theme_set(theme_classic(base_size=20)))+
theme(axis.text.x = element_text(angle = 45, hjust = .5, vjust = .5)) + #x轴label倾斜45度
guides(fill = guide_legend(title = NULL)) +
theme(legend.background = element_blank(), #移除整体边框
#图例的左下角置于绘图区域的左下角
legend.position=c(0,0),legend.justification = c(0,0))
p + geom_split_violin(data = tcga_gtex_MESO,
mapping = aes(x = tissue, y = tpm, fill = type2),
draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = "solid", #周围线的轮廓
color = "black", #周围线颜色
size = 0.2,
na.rm = T,
position ="identity") +
geom_split_violin(data = tcga_gtex_UVM,
mapping = aes(x = tissue, y = tpm, fill = type2),
draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = "solid", #周围线的轮廓
color = "black", #周围线颜色
size = 0.2,
na.rm = T,
position ="identity") +
scale_x_discrete(limits = levels(tcga_gtex$tissue))
ggsave("TP53expression.pdf",width = 20, height = 6)
 发表于 2025-9-8 07:44:24
发表于 2025-9-8 07:44:24
 |Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )
|Archiver|手机版|小黑屋|bioinfoer
( 萌ICP备20244422号 )